The GTF/GFF formats
The GTF/GFF formats are 9-column text formats used to describe and represent genomic features. The formats have quite evolved since 1997, and despite well-defined specifications existing nowadays they have a great flexibility allowing holding wide variety of information. This flexibility has a drawback aspect, there is an incredible amount of flavor of the formats: GFF / GFF1 / GFF2 / GFF2.5 / GFF3 / GTF / GTF2 / GTF2.1 / GTF2.2 / GTF2.5 / GTF3
It's often hard to understand and differentiate all GFF/GTF formats/flavors. Many tools using GTF/GFF formats fails due to specific expectations. Here is a comprehensive overview of the formats and their histories to help disentangle this complexity.
Table of Contents
Forewords
⇨ When I use the term gff it includes all gff formats/flavors. (The first version of the format was not called gff1 but gff. But to make it easier I will always specify the version of the format saying gff1 when I talk about the first version of it).
⇨ In the same way, when I use the term gtf it includes all gtf formats/flavors.
⇨ I have created the term gxf that means all the gff and gtf formats/flavors.
Introduction
GFF (Gene-Finding Format) is a general-purpose genome annotation format. It was conceived during a 1997 meeting on computational genefinding at the Isaac Newton Institute, Cambridge, UK and developed in collaboration between the Sanger Centre, the University of California at Santa Cruz and other participants (Holmes I: Studies in Probabilistic Sequence Alignment and Evolution. PhD thesis University of Cambridge 1998.). The key players have been Richard Durbin and David Haussler.
GFF is a one-line-per-record format that can be used to identify a substring of a biological sequence. It is an extension of a basic (name, start, end) tuple (or "NSE"). It was originally designed as a common format for sharing information between gene-finding sensors (finding signals: starts, splice sites, stops, motifs; finding regions: exons, introns, protein domains etc.), and gene predictors (predicting gene, RNA transcript or protein structures). These two steps were usually performed within the same program but thanks to the GFF format they can be decoupled, allowing the transfer of feature information from one tool to another. But its uses go beyond gene-finding and it is used as a convenient way to represent a set of many kinds of features. The GFF fomat has been developed to be easy to parse and process by a variety of programs in different languages (e.g Unix tools as grep and sort, perl, awk, etc.). For these reasons, they decided that each feature is described on a single line, and line order is not relevant.
GFF
GFF0
(before 13-11-1997)
There is no clear information about how the format looked at that time but it was close to the GFF1 format specification without the field source added the 1997-11-13.
GFF1
(13-11-1997)
For a complete description of the format please refer to this link: https://web.archive.org/web/19980222142332/http://www.sanger.ac.uk:80/~rd/gff.html. This is the oldest description of the format I found (1998-02-22).
I consider the format as GFF1 when they definitely defined the 9th field of the format (1997-11-13 rd: added extra source field as discussed at Newton Institute meeting 971029). Before that, the format only existed in its 0th version.
This GFF1 format contains 8 mandatory fields followed by one optional field. The fields are:
<seqname> <source> <feature> <start> <end> <score> <strand> <frame> [group]
They are defined as follows:
<seqname>
The name of the sequence. Having an explicit sequence name allows a feature file to be prepared for a data set of multiple sequences. Normally the seqname will be the identifier of the sequence in an accompanying fasta format file. An alternative is that 'seqname' is the identifier for a sequence in a public database, such as an EMBL/Genbank/DDBJ accession number. Which is the case, and which file or database to use, should be explained in accompanying information.
<source>
The source of this feature. This field will normally be used to indicate the program making the prediction, or if it comes from public database annotation, or is experimentally verified, etc.
<feature>
The feature type name. We hope to suggest a standard set of features, to facilitate import/export, comparison etc.. Of course, people are free to define new ones as needed. For example, Genie splice detectors account for a region of DNA, and multiple detectors may be available for the same site, as shown above.
<start>, <end>
Integers. <start> must be less than or equal to <end>. Sequence numbering starts at 1, so these numbers should be between 1 and the length of the relevant sequence, inclusive.
<score>
A floating point value. When there is no score (i.e., for a sensor that just records the possible presence of a signal, as "splice5" above) you must give something, by convention 0.
<strand>
One of '+', '-' or '.'. '.' should be used when strand is not relevant, e.g. for dinucleotide repeats.
<frame>
One of '0', '1', '2' or '.'. '0' indicates that the specified region is in frame, i.e., that its first base corresponds to the first base of a codon. '1' indicates that there is one extra base, i.e., that the second base of the region corresponds to the first base of a codon, and '2' means that the third base of the region is the first base of a codon. If the strand is '-', then the first base of the region is value of <end>, because the corresponding coding region will run from <end> to <start> on the reverse strand.
[group]
An optional string-valued field that can be used as a name to group together a set of records. Typical uses might be to group the introns and exons in one gene prediction (or experimentally verified gene structure), or to group multiple regions of match to another sequence, such as an EST or a protein. See below for examples.
=> All strings (i.e., values of the <seqname>, <feature> or <group> fields) should be under 256 characters long, and should not include whitespace. The whole line should be under 32k long. A character limit is not very desirable, but helps write parsers in some languages. The slightly silly 32k limit is to allow plenty of space for comments/extra data.
=> Fields must be separated by TAB characters ('\t').
Extra features of the format:
Comments
Comments are allowed, starting with "#" as in Perl, awk etc. Everything following # until the end of the line is ignored. Effectively this can be used in two ways. Either it must be at the beginning of the line (after any whitespace), to make the whole line a comment, or the comment could come after all the required fields on the line.
We also permit extra information to be given on the line following the group field without a '#' character. This allows extra method-specific information to be transferred with the line. However, we discourage overuse of this feature: better to find a way to do it with more true feature lines, and perhaps groups.
## comment lines for meta information
There is a set of standardised (i.e., parsable) ## line types that can be used optionally at the top of a gff file. The philosophy is a little like the special set of %% lines at the top of postscript files, used for example to give the BoundingBox for EPS files.
Current proposed ## lines are:
##gff-version 1
GFF version - in case it is a real success and we want to change it. The current version is 1.
##source-version {source} {version text}
So that people can record what version of a program or package was used to make the data in this file. I suggest the version is text without whitespace. That allows things like 1.3, 4a etc.
##date {date}
The date the file was made, or perhaps that the prediction programs were run. We suggest to use astronomical format: 1997-11-08 for 8th November 1997, first because these sort properly, and second to avoid any US/European bias.
##DNA {seqname}
##acggctcggattggcgctggatgatagatcagacgac
##...
##end-DNA
To give a DNA sequence. Several people have pointed out that it may be convenient to include the sequence in the file. It should not become mandatory to do so. Often the seqname will be a well-known identifier, and the sequence can easily be retrieved from a database, or an accompanying file.
##sequence-region {seqname} {start} {end}
To indicate that this file only contains entries for the the specified subregion of a sequence.
Please feel free to propose new ## lines. The ## line proposal came out of some discussions including Anders Krogh, David Haussler, people at the Newton Institute on 1997-10-29 and some email from Suzanna Lewis. Of course, naive programs can ignore all of these...
Here is an example of GFF1:
##gff-version 1
SEQ1 EMBL atg 103 105 . + 0
SEQ1 EMBL exon 103 172 . + 0
SEQ1 EMBL splice5 172 173 . + .
SEQ1 netgene splice5 172 173 0.94 + .
# this is comment that will be skipped by the parser
SEQ1 genie sp5-20 163 182 2.3 + .
SEQ1 genie sp5-10 168 177 2.1 + .
SEQ2 grail ATG 17 19 2.1 - 0
SEQ3 pred exon 100 135 . + 0 locus1 # this is also a comment that will be skipped by the parser
SEQ3 pred exon 235 260 . + 2 locus1 This is an example of extra information... They discourage overuse of this feature.
SEQ3 pred exon 360 396 . + 0 locus1
GFF2
(29-09-2000)
/!\ Note: Some of the changes we will see have been implemented before the offical release of GFF2. As a consequence, several interemediate states between version 1 and 2 have existed. We can call them GFF1.X. I will not further discuss these intermediate states.
16/12/98: Discussions with Lincoln Stein and others, the Version 2 format of GFF is proposed.
17/11/99: Gene Feature Finding Version 2 format is conceptually generalized to be the General Feature Format
The GFF2 format is conceptualized since the 16/12/98 but becomes officially the default version the 2000-9-29. Here is the official description which is a snapshot from here: https://web.archive.org/web/20010208224442/http://www.sanger.ac.uk:80/Software/formats/GFF/GFF_Spec.shtml.
You can find the first description (03 Feb 2000) of the GFF2 here that comes from here: ftp://ftp.sanger.ac.uk/pub/resources/software/gff-old/gff/.
Here we will review changes from GFF1.
=> The Gene Feature Finding has been generalized to accomodate RNA and Protein feature files and has been renamed the General Feature Format while retaining the same acronym GFF.
The main change from Version 1 to Version 2 is the revision of the optional 9th field with tag-value type structure (essentially semicolon-separated .ace format) used for any additional material on the line. Version 2 also allows '.' as a score, for features for which there is no score.
With the changes taking place to version 2 of the format, we also allow for feature sets to be defined over RNA and Protein sequences, as well as genomic DNA. This is used for example by the EMBOSS project to provide standard format output for all features as an option. In this case the
Definition
This GFF2 format contains 8 mandatory fields followed by optional fields. Fields are:
<seqname> <source> <feature> <start> <end> <score> <strand> <frame> [group/attributes] [comments]
Differences in these fields from GFF1 are as follows:
<seqname>
/
<source>
/
<feature>
Version 2 change: Standard Table of Features - we would like to enforce a standard nomenclature for common GFF features. This does not forbid the use of other features, rather, just that if the feature is obviously described in the standard list, that the standard label should be used. For this standard table we propose to fall back on the international public standards for genomic database feature annotation, specifically, the DDBJ/EMBL/GenBank feature table.
<start>, <end>
Version 2 change: version 2 condones values of <start> and <end> that extend outside the reference sequence. This is often more natural when dumping from acedb, rather than clipping. It means that some software using the files may need to clip for itself.
<score>
Version 2 change: When there is no score (i.e., for a sensor that just records the possible presence of a signal, as for the EMBL features above) you should use '.' instead of 0.
<strand>
Version 2 change: This field is left empty '.' for RNA and protein features.
<frame>
Version 2 change: This field is left empty '.' for RNA and protein features.
[group/attribute]
[New] Standard Table of Attribute Tag Identifiers The semantics of tags in attribute field tag-values pairs has not yet been completely formalized, however a useful constraint is that they be equivalent, where appropriate, to DDBJ/EMBL/GenBank feature 'qualifiers' of given features (see EMBL feature descriptions).
In addition to these, ACEDB typically dumps GFF with specific tag-value pairs for given feature types. These tag-value pairs may be considered 'standard' GFF tag-values with respect to ACEDB databases. (rbsk: These will be summarized in a table here in the near future)
Version 2 change: In version 2, the optional [group] field is renamed to [attribute] (09/99) and must have an tag-value structure following the syntax used within objects in a .ace file, flattened onto one line by semicolon separators. Tags must be standard identifiers ([A-Za-z][A-Za-z0-9_]*). Free text values must be quoted with double quotes. Note: all non-printing characters in such free text value strings (e.g. newlines, tabs, control characters, etc) must be explicitly represented by their C (UNIX) style backslash-escaped representation (e.g. newlines as '\n', tabs as '\t'). As in ACEDB, multiple values can follow a specific tag. The aim is to establish consistent use of particular tags, corresponding to an underlying implied ACEDB model if you want to think that way (but acedb is not required). Examples of these would be:
seq1 BLASTX similarity 101 235 87.1 + 0 Target "HBA_HUMAN" 11 55 ; E_value 0.0003
dJ102G20 GD_mRNA coding_exon 7105 7201 . - 2 Sequence "dJ102G20.C1.1"
=> Version 2 change: field and line size limitations are removed; however, fields (except the optional [attribute] field above) must still not include whitespace.
=> Version 2 note: previous Version 2 permission to use arbitrary whitespace as field delimiters is now revoked! (99/02/26)
Extra features of the format:
Comments
[...]
We also permit extra information to be given on the line following the attribute field without a '#' character (Version 2 change: this extra information must be delimited by the '#' comment delimiter OR by another tab field delimiter character, following any and all [attribute] field tag-value pairs).
[...]
Version 2 change: we gave in and defined a structured way of passing additional information, as described above under [attribute]. But the sentiment of this paragraph still applies - don't overuse the tag-value syntax. The use of tag-value pairs (with whitespace) renders problematic the parsing of Version 1 style comments (following the attribute field, without a '#' character), so in Version 2, such [attribute] trailing comments must either start with the "#" as noted above, or with at least one additional tab character. Moreover, '#' characters embedded within quoted text string values of [attribute] tag-values should not be parsed as the beginning of a comment.
## comment lines for meta information
There is a set of standardised (i.e., parsable) ## line types that can be used optionally at the top of a gff file. The philosophy is a little like the special set of %% lines at the top of postscript files, used for example to give the BoundingBox for EPS files.
Current proposed ## lines are:
##gff-version 2
The current version is 2. (Version 2 change!)
##source-version {source} {version text}
/
##date {date}
/
##Type <type> [<name>]
[New] The type of host sequence described by the features. Standard types are 'DNA', 'Protein' and 'RNA'. The optional <name> allows multiple ##Type definitions describing multiple GFF sets in one file, each which have a distinct type. If the name is not provided, then all the features in the file are of the given type. Thus, with this meta-comment, a single file could contain DNA, RNA and Protein features, for example, representing a single genomic locus or 'gene', alongside type-specific features of its transcribed mRNA and translated protein sequences. If no ##Type meta-comment is provided for a given GFF file, then the type is assumed to be DNA.
##DNA {seqname}
##acggctcggattggcgctggatgatagatcagacgac
##...
##end-DNA
/
##RNA <seqname>
##acggcucggauuggcgcuggaugauagaucagacgac
##...
##end-RNA
Similar to DNA. Creates an implicit ##Type RNA <seqname> directive.
##Protein <seqname>
##MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSF
##...
##end-Protein
Similar to DNA. Creates an implicit ##Type Protein <seqname> directive.
##sequence-region {seqname} {start} {end}
/
Here is an example of GFF2:
##gff-version 2
SEQ1 EMBL atg 103 105 . + 0
SEQ1 netgene splice5 172 173 0.94 + .
# this is comment that will be skipped by the parser
SEQ1 genie sp5-10 168 177 2.1 + .
SEQ2 grail ATG 17 19 2.1 - 0
SEQ3 BLASTX similarity 100 135 . + 0 Target "HBA_HUMAN" ; E_value 0.0003 # this is also a comment that will be skipped by the parser
SEQ3 BLASTX similarity 235 260 . + 2 Target "HBA_HUMAN" ; E_value 0.0005
SEQ3 BLASTX similarity 360 396 . + 0 Target "HBA_HUMAN" ; E_value 0.001
GFF3
(2004)
GFF3 addresses several shortcomings in its predecessor, GFF2. Actually it addresses the most common extensions to GFF, while preserving backward compatibility with previous formats. It has been conceptualized by Lincoln Stein. The First specification draft I found is from 10 September 2003 (version 1.00rc1). The first offical specification is the version 1 published the 30 September 2004.
Last updated the 26 February 2013 with version 1.23. Here is the last description of the specifications: https://github.com/The-Sequence-Ontology/Specifications/blob/master/gff3.md
The majors updates are:
the limitations of the feature type (3rd column) that is constrained to be either a term from the Sequence Ontology or an SO accession number (2278 possibilities). * Some attribute's tags have predefined meanings: ID, Name, Alias, Parent, Target, Gap, Derives_from, Note, Dbxref, Ontology_term, Is_circular.
Parent, Alias, Note, Dbxref and Ontology_term attributes can have multiple values (separated with the comma "," character).
The ID indicates the ID of the feature. The ID attribute is required for features that have children (e.g. gene and mRNAs), or for those that span multiple lines, but are optional for other features. IDs for each feature must be unique within the scope of the GFF file. In the case of discontinuous features (i.e., a single feature that exists over multiple genomic locations) the same ID may appear on multiple lines. All lines that share an ID must collectively represent a single feature.
The reserved Parent attribute can be used to establish a part-of relationship between two features. A feature that has the Parent attribute set is interpreted as asserting that it is a part of the specified Parent feature
GTF
GTF1
(2000 - generally called GTF)
GTF stands for Gene Transfer Format.
GTF borrows from the GFF file format [1], but has additional structure that warrants a separate definition and format name. The structure is similar to GFF, so the fields are:
<seqname><source><feature><start><end><score><strand><frame><attributes>
In this paper from 2003 (Keibler E, Brent M: Eval: a software package for analysis of genome annotations. BMC Bioinformatics 2003, 4:50.) they say:
Annotations are submitted to Eval in GTF file format http://genes.cse.wustl.edu/GTF2.html,
a community standard developed in the course of several collaborative genome annotations projects
[Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SE. Genome annotation assessment in
Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. | Mouse Genome
Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature.
2002;420:520–562. doi: 10.1038/nature01262.].
As such it can be run on the output of any annotation system.
So the oldest paper they point to is the one from Reese et al. from February 9, 2000 (Genome annotation assessment in Drosophila melanogaster) that says:
We found that the General Feature Format (GFF) (formerly known as the Gene Feature Finding
format) was an excellent fit to our needs. The GFF format is an extension of a simple name,
start, end record that includes some additional information about the sequence being annotated:
the source of the feature; the type of feature; the location of the feature in the sequence; and
a score, strand, and frame for the feature. It has an optional ninth field that can be used to
group multiple predictions into single annotations.
More information can be found at the GFF web site: http://www.sanger.ac.uk/Software/formats/GFF/.
Our evaluation tools used a GFF parser for the PERL programming language that is also available
at the GFF web site. We found that it was necessary to specify a standard set of feature names
within the GFF format, for instance, declaring that submitters should describe coding exons with
the feature name CDS.
From this, we can understand that in ~2000 for the drosophila genome project they have use the GFF format with particuliar specification that lead to the emergence of the GTF format.
In another paper, The Human Genome Browser at UCSC. Genome Res. 2002 Jun; 12(6): 996–1006. doi:10.1101/gr.229102 the authors say that the GTF format has been designed specifically for the human genome project:
Since August 2001, it has become possible for users to upload their own annotations for display
in the browser. These annotations can be in the standard GFF format
(http://www.sanger.ac.uk/Software/formats/GFF), or in some formats designed specifically for the
human genome project including GTF, PSL, and BED. The formats are described in detail in the web
page http://genome.cse.ucsc.edu/goldenPath/help/customTrack.html. Note that the GFF and GTF files
must be tab delimited rather than space delimited.
With this last paper, it's hard to understand from which project the GTF format is finally born.
Until now I havn't find a comprehensive description of the original GTF version (Ensembl version ?), but based on some hints from the Masters Project Report of Evan Keibler Eval: A Gene Set Comparison System and from other ressources I can say that the GTF1 was similar to GTF2. With few differences listed here:
feature
The feature field can take 5 values: `CDS`, `start_codon`, `stop_codon`, `exon` and `intron`.
strand
The strand value must be `+`, `-`, or `.`.
GTF2 and GFF2.5
(2003)
We can find in some places that GTF2 is similar to GFF2.5, but I do not know of a coherent explanation about it. It sounds definitely to be GTF format. The original GTF described by Ensembl has been adapted and became GTF2 for the need of the the Mouse/Human Annotation Collaboration.
Here the description from the Brent Lab at the Washington University in St. Louis. Found from the Eval publication received the 18 July 2003 mentioning the address http://genes.cse.wustl.edu/GTF2.html that has been archived in the web-archive the 12/12/2003. Prior to the publication in BMC Bioinformatics (and after 1 January 2003 because it's the most recent journal cited in his report) E. Kleiber released a Master project report named "Eval: A Gene Set Comparison System" where he mention and describe the GTF, maybe the first version of the GTF2 format.
GTF borrows from the GFF file format [1], but has additional structure that warrants a separate definition and format name. The structure is similar to GFF, so the fields are:
<seqname><source><feature><start><end><score><strand><frame><attributes>
Definition of these fields are:
<seqname>
The <seqname> field contains the name of the sequence which this gene is on.
<source>
The <source> field should be a unique label indicating where the annotations came from – typically the name of either a prediction program or a public database.
<feature>
The <feature> field can take 4 values: "CDS", "start_codon", "stop_codon" and "exon". The “CDS” feature represents the coding sequence starting with the first translated codon and proceeding to the last translated codon. Unlike Genbank annotation, the stop codon is not included in the “CDS” feature for the terminal exon. The “exon” feature is used to annotate all exons, including non-coding exons. The “start_codon” and “stop_codon” features should have a total length of three for any transcript but may be split onto more than one line in the rare case where an intron falls inside the codon.
<start>, <end>
Integer start and end coordinates of the feature relative to the beginning of the sequence named in <seqname>. <start> must be less than or equal to <end>. Sequence numbering starts at 1. Values of <start> and <end> must fall inside the sequence on which this feature resides.
<score>
The <score> field is used to store some score for the feature. This can be any numerical value, or can be left out and replaced with a period.
<strand>
'+' or '-'.
<frame>
A value of 0 indicates that the first whole codon of the reading frame is located at 5'-most base. 1 means that there is one extra base before the first whole codon and 2 means that there are two extra bases before the first whole codon. Note that the frame is not the length of the CDS mod 3. If the strand is '-', then the first base of the region is value of <end>, because the corresponding coding region will run from <end> to <start> on the reverse strand.
<attributes>
Each attribute in the <attribute> field should have the form: attribute_name “attribute_value”;
Attributes must end in a semicolon which must then be separated from the start of any subsequent attribute by exactly one space character (NOT a tab character). Attributes’ values should be surrounded by double quotes.
All four features have the same two mandatory attributes at the end of the record:
| qualifier | description |
|---|---|
| gene_id | A unique identifier for the genomic source of the transcript. Used to group transcripts into genes. |
| transcript_id | A unique identifier for the predicted transcript. Used to group features into transcripts. |
These attributes are designed for handling multiple transcripts from the same genomic region. Any other attributes or comments must appear after these two.
[comments].
Any line may contain comments. Comments are indicated by the # character and everything following a # character on any line is a comment. As such, all fields are prohibited from containing # characters
Here an example of GTF:
Hs-Ch1 Twinscan exon 150 200 . + . gene_id "1"; transcript_id "1.a";
Hs-Ch1 Twinscan exon 300 401 . + . gene_id "1"; transcript_id "1.a";
Hs-Ch1 Twinscan CDS 380 401 . + 0 gene_id "1"; transcript_id "1.a";
Hs-Ch1 Twinscan exon 501 650 . + . gene_id "1"; transcript_id "1.a";
Hs-Ch1 Twinscan CDS 501 650 . + 2 gene_id "1"; transcript_id "1.a";
Hs-Ch1 Twinscan exon 700 800 . + . gene_id "1"; transcript_id "1.a";
Hs-Ch1 Twinscan CDS 700 707 . + 2 gene_id "1"; transcript_id "1.a";
Hs-Ch1 Twinscan exon 900 997 . + . gene_id "1"; transcript_id "1.a";
Hs-Ch1 Twinscan start_codon 380 382 . + 0 gene_id "1"; transcript_id "1.a";
Hs-Ch1 Twinscan stop_codon 708 710 . + 0 gene_id "1"; transcript_id "1.a";
GTF2.1
(2005)
Here the description from the Brent Lab at the Washington University in St. Louis. In this version, the feature field change a little bit and can contain 6 different types:
The following feature types are required: "CDS", "start_codon", "stop_codon".
The features "5UTR", "3UTR", and "exon" are optional. All other features will be ignored.
GTF2.2
(2007)
In this version, they included specific 9th column terms: transcript_id, protein_id and gene_id.
Here the description from the Brent Lab at the Washington University in St. Louis.
The
The following feature types are required: "CDS", "start_codon", "stop_codon".
The features "5UTR", "3UTR", "inter", "inter_CNS", "intron_CNS" and "exon" are optional.
inter and inter_CNS should have an empty transcript_id and gene_id attribute: gene_id ""; transcript_id "";
GTF2.5
(2012)
This version is unofficial. I call it like that to differentiate it against the other GTF flavors. This GTF flavor has been developed by the GENCODE project. Here is the first desciption of this format and the last desciption of this format. The
The tags/key-name of the
GTF3
(2015)
This version is unofficial. I call it like that to differentiate it against the other GTF flavors. Originally Ensembl has created the GTF format that has been then slightly modified into GTF2 and then broadly used. Ensembl has adopted GTF2 and used only 4 different type of feature (CDS, exon, start_codon, stop_codon) and a lot of specific attributes. Then they adopted from release 75 the GTF 2.5. More features types are used: gene, transcript, exon, CDS, Selenocysteine, start_codon, stop_codon, and UTR. Then from release 82 they move on in the format I call GTF3 where they replaced UTR by three_prime_utr and five_prime_utr.
Resume
Timeline of the different formats
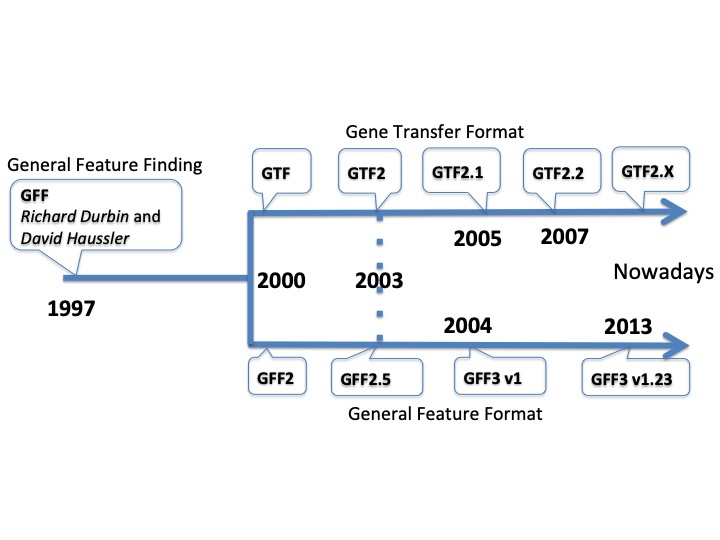
Main points and differences between GFF formats
| format version | year | col1 - seqname | col2 - source | col3 - feature | col4 - start | col5 - end | col6 - score | col7 - strand | col8 - frame | col9 - attribute | Comment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GFF1 | 1997 | can be anything | integer | integer | numerical value or 0 | '+', '-' or '.' | '0', '1', '2' or '.' | This field is originaly called group. An optional string-valued field that can be used as a name to group together a set of records. | Each String had to be under 256 characters + whole line under 32 000 characters long | ||
| GFF2 | 2000 | can be anything | integer | integer | numerical value or '.' | '+', '-' or '.' | '0', '1', '2' or '.' | This optional must have an tag value structure following the syntax used within objects in a .ace file, flattened onto one line by semicolon separators. Tags must be standard identifiers ([A-Za-z][A-Za-z0-9_]). Free text values must be quoted with double quotes. Note: all non-printing characters in such free text value strings (e.g. newlines, tabs, control characters, etc) must be explicitly represented by their C (UNIX) style backslash-escaped representation (e.g. newlines as '\n', tabs as '\t'). As in ACEDB, multiple values can follow a specific tag. form: Target "HBA_HUMAN" 11 55 ; E_value 0.0003* | The START and STOP codons are included in the CDS | ||
| GFF3 | 2004 | [a-zA-Z0-9.:^*$@!+_?-|] | Column name changed by |
integer | integer | numerical value or '.' | '+', '-', '.' or '?' | Column name changed by |
Multiple tag=value pairs are separated by semicolons. URL escaping rules are used for tags or values containing the following characters: ",=;". Spaces are allowed in this field, but tabs must be replaced with the %09 URL escape. Attribute values do not need to be and should not be quoted. The quotes should be included as part of the value by parsers and not stripped. form: ID=cds00004;Parent=mRNA00001,mRNA00002;Name=edenprotein.4. Some tags have predefined meaning, they start by capital letter. The ID attributes are only mandatory for those features that have children (the gene and mRNAs), or for those that span multiple lines. Consequently features having parents must have the Parent attribute. | The START and STOP codons are included in the CDS |
Main points and differences between GTF formats
| format version | year | col1 - seqname | col2 - source | col3 - feature | col4 - start | col5 - end | col6 - score | col7 - strand | col8 - frame | col9 - attribute | Comment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GTF1 | 2000 | CDS, start_codon, stop_codon, exon, intron | integer | integer | numerical value or '.' | '+', '-' or '.' | '0', '1', '2' or '.' | porbably similar to GTF2 | probably similar to GTF2 | ||
| GTF2 / GFF2.5 | 2003 | CDS, start_codon, stop_codon, exon | integer | integer | numerical value or '.' | '+' or '-' | '0', '1', '2' or '.' | Attributes must end in a semicolon which must then be separated from the start of any subsequent attribute by exactly one space character (NOT a tab character). Attributes’ values should be surrounded by double quotes. form: attribute_name “attribute_value”; attribute_name “attribute_value”; Two mandatory attributes: gene_id, transcript_id. Any other attributes or comments must appear after these two and will be ignored. Textual attributes should be surrounded by doublequotes. | Unlike Genbank annotation, the stop codon is not included in the CDS for the terminal exon | ||
| GTF2.1 | 2005 | CDS, start_codon, stop_codon, exon, 5UTR, 3UTR | integer | integer | numerical value or '.' | '+' or '-' | '0', '1', '2' or '.' | Attributes must end in a semicolon which must then be separated from the start of any subsequent attribute by exactly one space character (NOT a tab character). Attributes’ values should be surrounded by double quotes. form: attribute_name “attribute_value”; attribute_name “attribute_value”; Two mandatory attributes: gene_id, transcript_id. Any other attributes or comments must appear after these two and will be ignored. Textual attributes should be surrounded by doublequotes. | Unlike Genbank annotation, the stop codon is not included in the CDS for the terminal exon | ||
| GTF2.2 | 2007 | CDS, start_codon, stop_codon, 5UTR, 3UTR, inter, inter_CNS, intron_CNS and exon | integer | integer | numerical value or '.' | '+' or '-' | '0', '1', '2' or '.' | Attributes must end in a semicolon which must then be separated from the start of any subsequent attribute by exactly one space character (NOT a tab character). Attributes’ values should be surrounded by double quotes. form: attribute_name “attribute_value”; attribute_name “attribute_value”; Two mandatory attributes: gene_id, transcript_id. Any other attributes or comments must appear after these two and will be ignored. Textual attributes should be surrounded by doublequotes. | Unlike Genbank annotation, the stop codon is not included in the CDS for the terminal exon | ||
| GTF2.5 | 2012 | gene, transcript, exon, CDS, UTR, start_codon, stop_codon, Selenocysteine | integer | integer | numerical value or '.' | '+' or '-' | '0', '1', '2' or '.' | Attributes must end in a semicolon which must then be separated from the start of any subsequent attribute by exactly one space character (NOT a tab character). Attributes’ values should be surrounded by double quotes. form: attribute_name “attribute_value”; attribute_name “attribute_value”; ~9 mandatory tags but this number varies depending of the version and the type of feature. Number of optional tag varies between 34 and 82 depending of the version. Textual attributes should be surrounded by doublequotes. | Unlike Genbank annotation, the stop codon is not included in the CDS for the terminal exon | ||
| GTF3 | 2015 | gene, transcript, exon, CDS, Selenocysteine, start_codon, stop_codon, three_prime_utr and five_prime_utr | integer | integer | numerical value or '.' | '+' or '-' | '0', '1', '2' or '.' | Attributes must end in a semicolon which must then be separated from the start of any subsequent attribute by exactly one space character (NOT a tab character). Attributes’ values should be surrounded by double quotes. form: attribute_name “attribute_value”; attribute_name “attribute_value”; Two mandatory attributes: gene_id, transcript_id. Any other attributes or comments must appear after these two and will be ignored. Textual attributes should be surrounded by doublequotes. | Unlike Genbank annotation, the stop codon is not included in the CDS for the terminal exon |
Discussion
The main differences between GTF and GFF formats are the 3rd and 9th colomn. The feature type value of the 3rd column in GTF is constrained by a list of few feature types (<10) while in GFF it is much more vast. It was not constrained until version 3 where it is now constrained to be either a term from the Sequence Ontology or an SO accession number ( 2278 possible terms ).
The structure of the 9th column is slightly different between the two formats:
GTF2.2: attribute_name “attribute_value”; attribute_name “attribute_value”;
GFF3: ID=cds00004;Parent=mRNA00001,mRNA00002;Name=edenprotein.4
Within that column, the mandatory attributes are different.
Feature type limitation wihtin GTF
As the feature types of the 3rd column is limited by the GTF format, many groups/infrastructure use the 9th column to describe other features like tRNA, pseudogenes, etc. As an example, ENSEMBL use the attribute gene_biotype to define if a transcript feature is coding or non-coding.
Extra
Problems encountered due to lack of standardization
Inconsistency in stop codon treatment in GTF tracks (from https://genome.ucsc.edu/FAQ/FAQtracks.html):
I've been doing some comparative gene set analysis using the gene annotation tracks and I believe I have run into an inconsistency in the way that stop codons are treated in the annotations. Looking at the Human June 2002 assembly, the annotations for Ensembl, Twinscan, SGP, and Geneid appear to exclude the stop codon in the coding region coordinates. All of the other gene annotation sets include the stop codon as part of the coding region. My guess is that this inconsistency is the result of the gene sets being imported from different file formats. The GTF2 format does not include the stop codon in the terminal exon, while the GenBank format does, and the GFF format does not specify what to do.
Answer:
Your guess is correct. We haven't gotten around to fixing this situation. A while ago, the Twinscan group made a GTF validator. It interpreted the stop codon as not part of the coding region. Prior to that, all GFF and GTF annotations that we received did include the stop codon as part of the coding region; therefore, we didn't have special code in our database to enforce it. In response to the validator, Ensembl, SGP and Geneid switched their handling of stop codons to the way that Twinscan does it, hence the discrepancy.
Inconsistency in GTF format (reported by Evan Keibler in his Masters Project Report):
Although the GTF file format is a fairly simple and well defined format, data is often claimed to be in GTF format when it does not comply completely with the specification. Most data is generated in some proprietary format specific to the particular program or lab which produced it. These proprietary formats often differ in small subtle ways, such as the sequence being indexed starting at position 0 or 1, or the start/stop codon being inside or outside of the initial/terminal exon. If the data is to be effectively shared with others it must be in a standard, well defined format. Though many labs do convert their data to GTF format, the files they generate rarely comply completely with the specification. For this reason the GTF validator was created. The validator allows the user to verify that the data is in correct GTF format before sharing with others. This makes communication more efficient because the receiver does not have to locate and fix the subtle differences between the many file formats.
Ensembl GTF formats
Evolution of the 3rd and 9th column
Here the example of the human annotation:
| annotation file | ensembl version | nb feature type (3rd column) | nb attribute tag (9th column) |
|---|---|---|---|
| Homo_sapiens.NCBI36.43.gtf | 43 | 4: CDS exon start_codon stop_codon |
6: exon_number gene_id gene_name protein_id transcript_id transcript_name |
| Homo_sapiens.GRCh37.74.gtf | 74 | 4: CDS exon start_codon stop_codon |
8: exon_id exon_number gene_biotype gene_id gene_name protein_id transcript_id transcript_name |
| Homo_sapiens.GRCh37.75.gtf | 75 | 8: CDS Selenocysteine UTR exon gene start_codon stop_codon transcript |
12: ccds_id exon_id exon_number gene_biotype gene_id gene_name gene_source protein_id tag* transcript_id transcript_name transcript_source |
| Homo_sapiens.GRCh37.81.gtf | 81 | 8: CDS Selenocysteine UTR exon gene start_codon stop_codon transcript |
22: ccds_id exon_id exon_number exon_version gene_biotype gene_id gene_name gene_source gene_version havana_gene havana_gene_version havana_transcript havana_transcript_version protein_id protein_version tag* transcript_biotype transcript_id transcript_name transcript_source transcript_support_level transcript_version |
| Homo_sapiens.GRCh37.82.gtf | 82 | 9: CDS Selenocysteine exon five_prime_utr gene start_codon stop_codon three_prime_utr transcript |
22: same as before |
| Homo_sapiens.GRCh38.95.gtf | 95 | 9: CDS Selenocysteine exon five_prime_utr gene start_codon stop_codon three_prime_utr transcript |
18: ccds_id exon_id exon_number exon_version gene_biotype gene_id gene_name gene_source gene_version protein_id protein_version tag* transcript_biotype transcript_id transcript_name transcript_source transcript_support_level transcript_version |
| *Tags are additional flags used to indicate attibutes of the transcript. |
Difference between GENCODE and Ensembl GTF
From here.
The gene annotation is the same in both files. The only exception is that the genes which are common to the human chromosome X and Y PAR regions can be found twice in the GENCODE GTF, while they are shown only for chromosome X in the Ensembl file.
In addition, the GENCODE GTF contains a number of attributes not present in the Ensembl GTF, including annotation remarks, APPRIS tags and other tags highlighting transcripts experimentally validated by the GENCODE project or 3-way-consensus pseudogenes (predicted by Havana, Yale and UCSC). See our complete list of tags for more information.
Please note that the Ensembl GTF covers the annotation in all sequence regions whereas GENCODE produces a similar file but also a GTF file with the annotation on the reference chromosomes only.